Computing Gene Therapy Transduction Efficiencies
So this is going to be a long one, culminating from months of hard work. This was super fun to work on, and we truly believe simpler computational models solve and/or capture more intuitive insights from biological data.
Adeno-Associated Viruses (AAVs) have revolutionized gene therapy by acting as efficient delivery vehicles for genetic payloads. However, the diverse cell types that naturally occurring AAVs infect can lead to off-target effects, complicating treatments. The key to successful gene therapy lies in engineering AAVs that target only the intended cells with pinpoint accuracy.
In this post, we explore a data-driven statistical approach to predict AAV tropism based on the sequence of its surface protein, VP1. Using experimental data on transduction efficiencies, our pipeline predicts how effectively AAVs bearing specific VP1 sequences transduce various cell types. Impressively, the model achieves an R² accuracy of 92% for canonical AAVs and related variants.
This predictive capability represents a significant leap forward, offering researchers a faster, more reliable way to evaluate AAV candidates for targeted gene therapy. Join us as we dive into the details of this approach and its potential to transform AAV engineering for precise, effective treatments.
Gene Therapy: Rewriting the Blueprint of Life
Gene therapy is a groundbreaking approach that offers the possibility of curing genetic disorders by targeting the root cause—a faulty gene. At its core, gene therapy involves replacing a mutant, disease-causing gene with its healthy, wild-type counterpart. The goal? To restore normal gene expression and, in doing so, eliminate the underlying cause of the disorder.
While the concept sounds simple, the execution is anything but. Delivering a functional gene into the cells of a living human and ensuring it integrates into the genome is a monumental challenge. It requires not only a deep understanding of genetics but also cutting-edge tools for precise and effective delivery.
In most cases, the delivery vehicle, or vector, is a virus that has been repurposed for therapeutic use. The virus’s natural genetic material is replaced with the desired gene sequence, along with regulatory elements that facilitate its integration into the host genome. Once introduced into the body, the modified virus seeks out specific cells, gains entry using cellular receptors, and delivers the genetic material. The corrected gene is then incorporated into the patient’s DNA, setting the stage for healthy gene function.
This process holds immense promise but also presents significant technical and ethical challenges. Selecting the right vector, ensuring precise targeting, and safeguarding against unintended consequences are all critical considerations.
So what is the delivery vehicle or virus?
Adeno-associated viruses (AAVs) have emerged as one of the most widely used vectors for gene therapy, thanks to their small, ~5 kb single-stranded DNA genome and non-pathogenic nature in humans. Their viral capsid is primarily composed of the structural protein VP1, along with its truncated versions, VP2 and VP3, all derived from the same gene. These surface proteins are key players in mediating AAV entry into host cells by interacting with cellular receptors. Based on natural variations in the VP1 sequence, AAVs are categorized into serotypes, with the most commonly known being AAV1 through AAV9.
The versatility and safety profile of AAVs have made them ideal candidates for gene therapy, with numerous clinical trials and experimental treatments over the last decade demonstrating their promise. However, significant technical challenges remain, particularly in achieving precise targeting and avoiding off-target effects.
A major hurdle is the tropism of AAVs—their transduction efficiency (TE) across different cell types. This is a measure of the success of an AAV to penetrate a given cell type. Effective gene therapy often requires targeting specific cell types linked to a disease phenotype, but wild-type AAVs show variable tropism, infecting multiple tissues with differing efficiencies. Tropism is the characteristic we want to associate with high transduction efficiencies. While different AAV serotypes exhibit distinct tropisms, naturally occurring variants rarely provide the specificity required for high-efficiency delivery to a single target tissue with minimal off-target effects.
To overcome these limitations, engineering AAVs with enhanced specificity has become a critical focus. Developing tailored vectors with optimized tropism for precise cell targeting holds the key to advancing gene therapy, improving therapeutic outcomes, and minimizing unintended effects.

Natural affinities of different AAVs to different tissue types (Issa, S.S.; Shaimardanova, A.A.; Solovyeva, V.V.; Rizvanov, A.A. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells 2023, 12, 785. https://doi.org/10.3390/cells12050785)
Designing AAVs
Experimental approaches to creating AAVs with precise tropism have shown promise but often come with significant challenges. Methods like directed evolution involve iterative rounds of infection, extraction, and reinfection within specific tissues of model organisms or in cultured mammalian cells. Other strategies, such as rational design, introduce targeted changes into the viral protein sequence to alter its behaviour. While effective, these approaches are labour-intensive, time-consuming, and costly, highlighting the need for more efficient alternatives.
One such alternative is leveraging computational tools to predict AAV tropism based on the sequence of its surface proteins, particularly VP1. Since these surface proteins interact with host cell receptors and play a pivotal role in determining transduction efficiency (TE), analysing their sequence provides a logical starting point. By combining experimentally derived TE data for various AAV serotypes with VP1 sequence information, it becomes possible to develop predictive models. These models offer a streamlined approach to identifying candidate AAV sequences with desired tropism, significantly reducing the experimental workload and accelerating the development of specific AAVs for gene therapy.
Computational Workflow
Proof of concept

Prediction of transduction efficiency

Proof of Concept
VP1 Sequence Similarity Mirrors Tropism Patterns
To predict the tropism of an AAV based on its VP1 protein sequence, we first needed to determine if transduction efficiency is indeed encoded in the sequence. To explore this, we analyzed the similarity between pairs of AAV serotypes using two approaches:
1. Transduction Efficiency Metrics: We utilized experimentally determined transduction efficiencies across various human cell types and visualized the data using principal component analysis (PCA).
2. Sequence Similarity Metrics: We calculated the number of shared k-mers (k = 8) between VP1 sequences from different serotypes as a measure of sequence similarity (heatmap below).
We then assessed whether the two metrics—based on transduction efficiency and sequence similarity—aligned with each other. If so, it would indicate that VP1 sequence similarity correlates with patterns of tropism, supporting the use of sequence data to predict AAV tropism.
The results were clear: the tropism patterns observed in the PCA plot matched the sequence similarity patterns derived from shared k-mers. For example, AAV1 and AAV6 showed strong similarity in both sequence and tropism, while AAV2 and AAV3 were more similar to each other than to other serotypes. Conversely, AAV1 and AAV2 were highly distinct by both metrics.
These consistent patterns validate the hypothesis that VP1 sequence similarity is a reliable proxy for predicting tropism, providing a solid foundation for developing sequence-based models to predict AAV tropism with unknown sequences.

Experimentally determined transduction efficiencies of various AAV serotypes for different human cell types (adapted from Ellis et al., 2013 Fig. 2). This dataset is used for the PCA below.


Predicting AAV Transduction Efficiencies
Building on the validation of our approach to predict transduction efficiency from VP1 sequence, we turned our focus to classifying existing AAV serotypes based on their known transduction efficiencies. Specifically, for each human cell type, we aimed to group the nine AAV serotypes into two distinct clusters based on their relative efficiency in transducing that cell type.
To achieve this, we applied k-means clustering (k = 2) to the transduction efficiency data for AAV1–9 for each cell type. This allowed us to identify patterns of similarity and distinctiveness in how different serotypes performed across cell types. The clustering results for each cell type are visualized below showcasing how the serotypes segregate into high- and low-efficiency clusters for each context.

Matrix showing which serotypes belong to the two clusters HigherTE and LowerTE for each cell type. The lighter shade represents HigherTE while the darker shade represents LowerTE.
Next, we calculated the mean transduction efficiency (TE) for each k-means cluster. The cluster with the higher mean TE was labeled as the "HigherTE" cluster, while the one with the lower mean TE was termed the "LowerTE" cluster. To ensure meaningful distinctions between clusters, we visually inspected the data and filtered out cell types where the TE differences between the two clusters were not clearly distinct. The figure below illustrates the transduction efficiencies of the HigherTE (red) and LowerTE (blue) clusters for the cell types that passed this filtering step.
Interestingly, several cell types exhibited identical clustering patterns. For example, both Keratinocyte and Jurkat cells showed the same grouping: HigherTE = {AAV1, AAV6} and LowerTE = {AAV2, AAV3, AAV4, AAV5, AAV7, AAV8, AAV9}. This raised an important question: should these cell types, despite having identical clusters, be treated as distinct in further analyses of TE prediction?
Figure below provides clarity. Even though the clusters themselves are identical, the transduction efficiencies of the HigherTE and LowerTE clusters vary between these cell types. This indicates that, while the patterns of clustering are similar, the underlying TE dynamics are unique to each cell type. As a result, we treated these cell types as distinct in our subsequent analyses of TE prediction based on AAV VP1 sequences.

Transduction efficiencies of AAV serotypes belonging to HigherTE (red) and LowerTE (blue) clusters for each cell type. The clusters were obtained by k-means clustering (k = 2).
Creating Multiple Sequence Alignments and Profile HMMs
With the HigherTE and LowerTE clusters identified for each cell type, we conducted multiple sequence alignments (MSA) for the VP1 sequences within the HigherTE cluster. For this, we utilized the MAFFT algorithm, implemented via the Jalview software, to ensure accurate and efficient alignments.
From these alignments, we constructed profile hidden Markov models (HMMs) for the HigherTE cluster of each cell type. These HMMs represent canonical profiles of VP1 sequences associated with higher transduction efficiency for the given cell type, serving as a reference for comparison against the LowerTE cluster sequences. This approach establishes a foundational model for understanding the sequence characteristics linked to efficient transduction in specific cell types.
In addition to the profile HMM for HigherTE, we also performed multiple sequence alignments of VP1 sequences belonging to each AAV serotype, i.e. nine MSAs, one each for AAV1 through AAV9. For each of these serotype alignments, we created a serotype profile HMM (see below for a schematic representation). We aimed to use these profiles for alignment against a query VP1 sequence to predict its transduction efficiency, as explained in subsequent sections.

Schematic representation of clusters HigherTE and LowerTE for an example cell type. Each cluster is made up of multiple serotypes. Each serotype in turn has multiple VP1 sequences belonging to it. We performed multiple sequence alignments (MSA) of (a) all sequences in the HigherTE cluster, and (b) all sequences of each serotype. We then generated profile HMMs from each MSA to get a HigherTE cluster profile for each cell type, and serotype profiles for each serotype AAV1 through AAV9.
Establishing a Null Score Distribution
To evaluate whether a given VP1 sequence with unknown transduction efficiency (TE) aligns significantly with the HigherTE cluster of a cell type, we calculated the Viterbi probability of the sequence against the profile HMM of the HigherTE cluster. This probability served as a key statistic to assess the alignment's significance.
To establish the null distribution of scores for each cell type, we evaluated sequences from the LowerTE cluster against the HigherTE profile HMM. Plotting these scores as a histogram revealed a multimodal Gaussian distribution (fig below, left). To simplify the analysis, we focused on the rightmost portion of the distribution, reasoning that if a sequence scores significantly relative to this subset, it would also be significant compared to the entire distribution.
To isolate this rightmost segment, we applied a Gaussian Mixture Model (GMM) to cluster the scores into distinct components. From this clustering, we identified the component representing the highest scores (Cluster 2, right). This subset of scores formed the basis of the null distribution, which we then fit to a normal distribution. By doing so, we established a robust framework for determining significant alignment scores, allowing for more precise classification of sequences with unknown TEs.

(Left) The histogram of scores of sequences in the LowerTE cluster when aligned to the HigherTE profile for a representative cell type. There are two normal distributions that the data is divided into that are apparent in the figure.
(Right) GMM (Gaussian Mixture Model) clustering of the ES-cell data. Two clusters representing the normal distributions that we saw in the figure on the left, form with distinct ranges of scores. The cluster in green was picked for statistical analysis as it represented the higher mean score. This was repeated for all cell types.

Fitted normal curve on the data points belonging to ‘Cluster 2’ shown previously for the representative cell type. The dashed red line shows the critical value, the magenta line represents the region on the curve that is significant, while the green line represents the region on the curve that is not significant. Similar distributions and Gaussian fits for all cell types.
We then test the null hypothesis: The given sequence with unknown TE belongs to the set of LowerTE sequences that formed the null distribution. To determine the significance of the Viterbi probability score we just calculated, we performed a one-tailed test at a 5% significance level. We classified a sequence as belonging to the HigherTE cluster if its alignment score was significant, or as belonging to the LowerTE cluster if the alignment score was not significant.
Predicting Transduction Efficiency
To predict the transduction efficiency (TE) of a given sequence (`Seq`) for each cell type, we take the following approach:
1. Alignment and Scoring: For each cell type, the sequence is aligned to the HigherTE and LowerTE profiles using the Viterbi algorithm, yielding corresponding probability scores.
2. Significance Testing: The alignment to the HigherTE profile is tested for significance using the method outlined earlier.
If the sequence produces a significant hit for a specific cell type (e.g., cell type X), we further refine the prediction. Assume the HigherTE profile for cell type X is composed of serotypes S1, S2, S3, and S4. The sequence is aligned individually with the set of sequences from each serotype, producing Viterbi probabilities p₁, p₂, p₃, and p₄. A weighted average of these probabilities is then calculated to determine the predicted TE for this cell type (formula below). Each serotype is assigned a single TE value derived from published experimental data, which is uniformly applied to all sequences associated with that serotype (identified via BLAST).
If no significant alignment to a HigherTE profile is detected for a cell type, the sequence is aligned to the serotypes in the LowerTE cluster, and the process is repeated to estimate the TE.
In cases where the sequence is significant for multiple cell types, transduction efficiencies are calculated for each significant cell type, providing a comprehensive profile of the sequence’s potential TE across various cell types.

An example calculation for Seq with an unknown TE value - considering [S1, S2, S3, S4] to be the four serotypes in a representative HigherTE cluster, and [p1, p2, p3, p4] to be the Viterbi probabilities associated with aligning Seq to the serotype profiles of S1, S2, S3, and S4 respectively, the predicted TE is a weighted average of the probability and corresponding experimentally determined TE values [TE1, TE2, TE3, TE4] for the serotypes, as shown by the above expression.
Validation of Predicted Transduction Efficiencies
To evaluate the performance of our pipeline for predicting transduction efficiency (TE) from AAV VP1 sequences, we validated the model using sequences from our original dataset, which included all nine serotypes. While these sequences were part of the training data used to build the profile HMMs, the impact of any single sequence on the overall predictions was minimal, making this a reasonable first step in validation. Each sequence was individually run through the pipeline to predict its TE, simulating a basic validation process. In future work, a more rigorous leave-one-out validation approach could provide a stronger assessment of the pipeline’s accuracy (see Discussion and Future Directions).
For each sequence, we compared the predicted TE against the experimental TE values for its corresponding serotype (from Ellis et al., 2013). These experimental values, represented by the respective serotype's column shown earlier in this post, were treated as the "true" transduction efficiencies for validation purposes. This approach assumes that all VP1 sequences within a given serotype have identical TEs, which limits our ability to account for sequence variation within serotypes.
While this simplification is not ideal, it was necessary due to the limitations of the available dataset. The Ellis et al. (2013) study remains the only comprehensive source of experimental TE data, but it does not provide the exact sequences for the serotypes used. As such, this method represents the most practical validation framework given the constraints of the data.
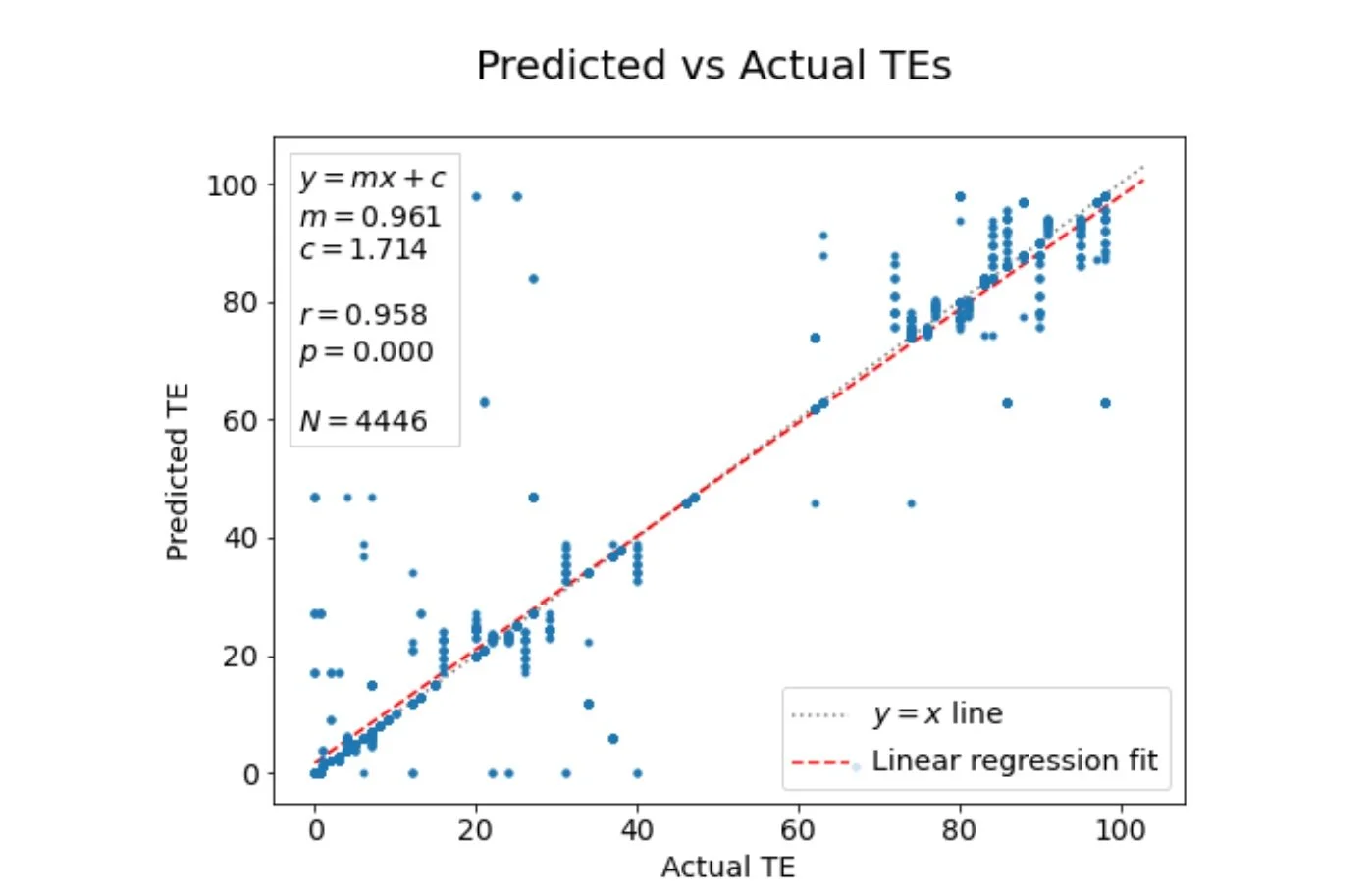
Scatterplot showing predicted transduction efficiencies (y-axis) vs the corresponding experimentally determined true transduction efficiencies (x-axis). The values of slope (m) and intercept (c) for the best fit linear regression, and the Pearson correlation coefficient (r) are indicated. Each AAV sequence in the database corresponds to 13 points on this plot, one for each cell type. We see a very good agreement between the predicted and true TEs, indicating that our approach works well.
Evaluating Model Performance
Our model demonstrates strong performance in predicting experimental transduction efficiencies across a range of AAV serotypes and cell types. With these results in hand, we sought to position our methodology within the broader context of AAV engineering.
Recent years have seen a surge in machine learning (ML)-based approaches to predict AAV capsid viability from protein or DNA sequence data (e.g., Bryant et al., 2021; Marques et al., 2021; Becker et al., 2022). Notably, a recent PhD thesis proposed an ML model to predict *in vivo* AAV tropisms in mice based on transduction efficiencies measured *in vitro* (Chen, 2020). However, to our knowledge, no published method has yet focused on directly predicting AAV transduction efficiencies from sequence information alone (Becker et al., 2022).
The field appears to be moving toward using ML not just to predict capsid viability but also for tropism prediction, as exemplified by recent preprints like Portell et al. (2022). As such models become available, they will provide valuable benchmarks for comparison with our approach.
Ultimately, the most definitive validation of our model would involve experimental testing of predicted transduction efficiencies for AAVs with specific VP1 sequences. This step would offer the clearest insight into how well our model translates from computational predictions to real-world applications.
Enhancing AAV Tropism Prediction: Performance, Limitations, and Future Directions
Performance Variability Across Serotypes and Cell Types
Our pipeline shows robust performance in predicting transduction efficiencies (TE) for most AAV serotypes and cell types. However, variability exists. For instance, AAV4 performed poorly, likely due to its small dataset (only two sequences), where even a single incorrect prediction significantly skews results. Similarly, AAV7 and AAV8 showed lower performance, potentially because some AAV7 sequences shared more similarity with AAV8 than within their own serotype. Improved experimental TE data capturing within-serotype variation could address these issues.
Across cell types, TE prediction accuracy also varied. For BJ-Fibroblasts and BJ-hTERT-Fibroblasts, performance was weaker, potentially due to the presence of AAV2-3 hybrid sequences, which are harder to classify accurately. In these cell types, AAV2 belongs to the HigherTE cluster, while AAV3 lies in LowerTE, unlike other cell types where they cluster together. Despite these challenges, for most cell types, the correlation coefficient \( r \) ranged between 0.84 and 0.96, with regression slopes close to unity, demonstrating overall strong performance.
Opportunities to Improve Preprocessing
While the pipeline performs well, improvements to the preprocessing steps could enhance accuracy. The current method relies on k-means clustering to divide VP1 sequences into HigherTE and LowerTE groups for each cell type. Since k-means can bias toward equal-sized clusters, alternative approaches like k-means++ or Gaussian mixture models (GMMs) could yield better-defined clusters.
Additionally, we currently visually assess the relationship between experimental TE patterns (via PCA) and sequence similarity (via shared k-mers). A more rigorous comparison using hierarchical clustering could provide a quantitative measure of how well sequence features explain tropism variability, enhancing the robustness of the pipeline.
Data Limitations and Potential Solutions
A significant challenge lies in the limited availability of experimental and sequence data. For some serotypes, such as AAV4 and AAV5, only a few sequences are publicly available, limiting the model’s ability to account for sequence diversity. Additionally, the most comprehensive TE dataset (Ellis et al., 2013) lacks associated VP1 sequences, preventing incorporation of within-serotype variation into validation. Expanding datasets with more diverse and annotated sequences, along with experimental TE values, would improve the null distribution fits and overall pipeline performance.
Another limitation is the reliance on *in vitro* TE data, which may not always translate to *in vivo* conditions. Direct use of *in vivo* TE datasets, where available, or incorporating models that map *in vitro* data to *in vivo* predictions, could bridge this gap and increase clinical relevance.
Improving Validation and Model Comparisons
Our current validation approach, which uses training data sequences for testing, introduces potential biases. Future validation steps should employ hold-one-out or hold-few-out methods to assess generalizability. Alternatively, the pipeline could be compared against emerging machine learning models that predict TE from protein sequences. Ultimately, experimental validation of predicted TEs for AAVs with specific VP1 sequences, in both *in vitro* and *in vivo* systems, would provide the most robust assessment of pipeline accuracy.
Designing AAVs with High Specificity
The ability to predict tropism opens new avenues for engineering AAVs with highly specific targeting. By integrating our pipeline with optimization algorithms like genetic algorithms (GAs), VP1 sequences could be iteratively designed to maximize specificity for one cell type while minimizing off-target effects. Alternatively, computationally generated VP1 sequences with extensive variation could be screened using our predictor, scoring for specificity to identify candidates with desirable tropism profiles.
Combining our approach with existing methods for generating and assessing AAV sequence viability offers a powerful toolset for designing targeted delivery vectors. Such advances in AAV engineering could transform gene therapy, enabling next-generation therapeutics with unparalleled precision for treating genetic disorders.